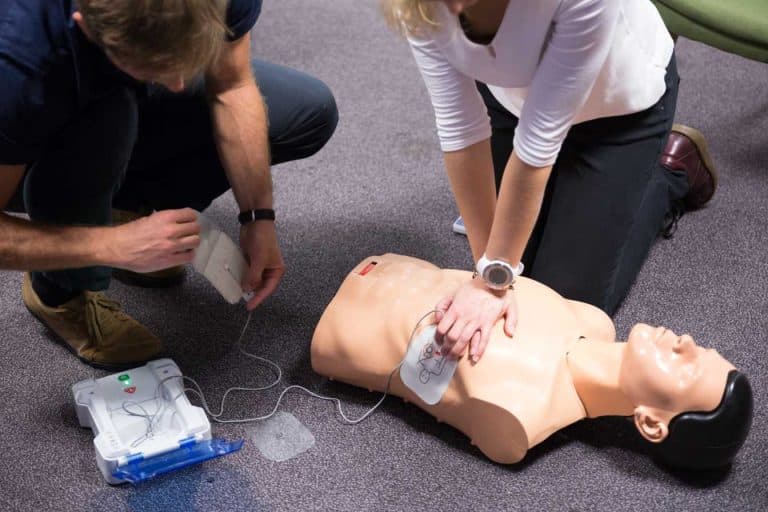
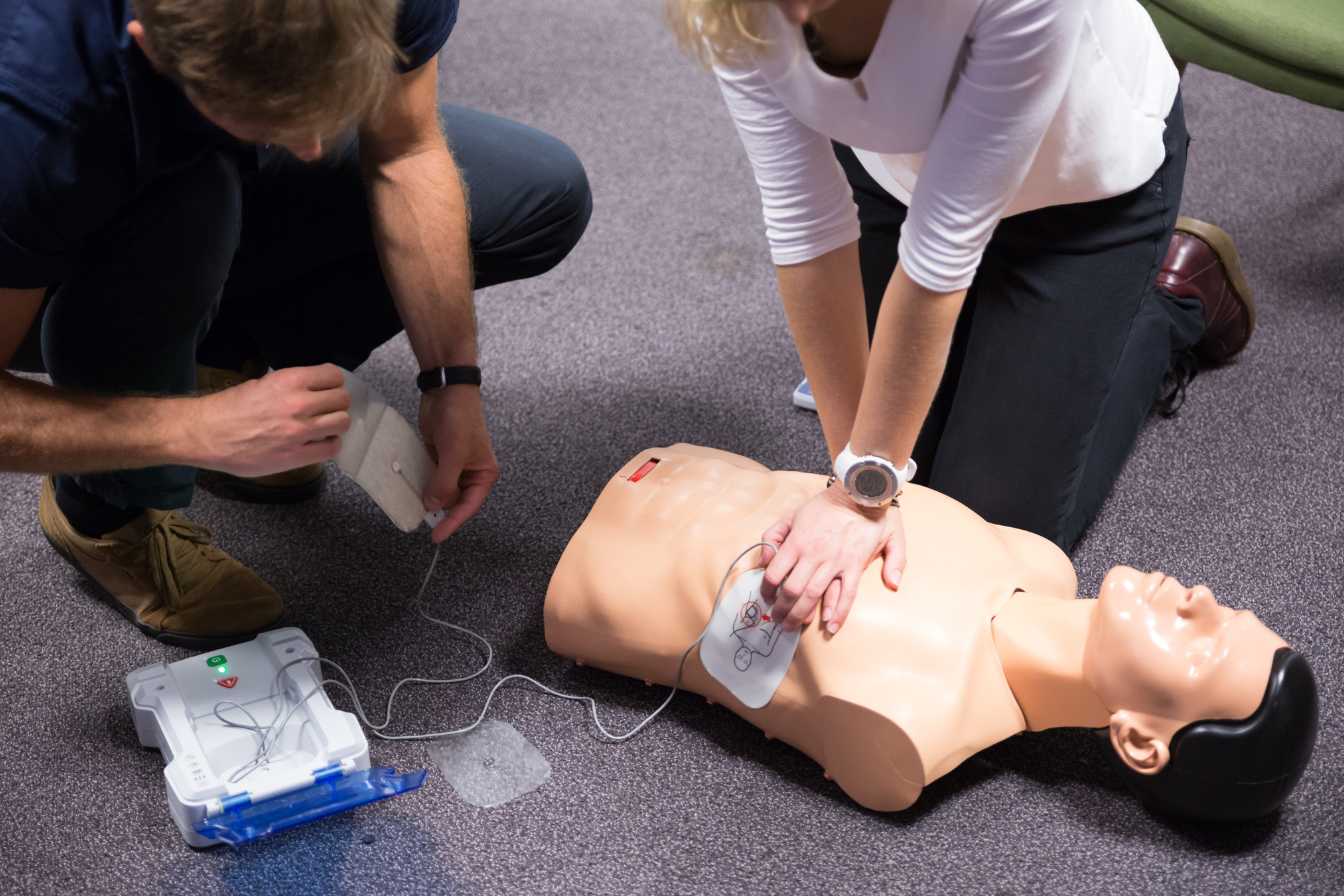
Depuis le 5 mai 2017, les défibrillateurs automatisés externes ont été reclassés en dispositif médical de classe III. Quels impacts pour les fabricants et exploitants ?
Quelle est la réglementation applicable sur les dispositifs médicaux en Europe ?
Les dispositifs médicaux sont régis par la directive 93/42/CEE. La directive reprend les exigences en matière de conformité, de normes harmonisées, de classification des dispositifs médicaux, … sa finalité étant de garantir la performance et la sécurité des dispositifs médicaux commercialisés sur le marché Européen.
Pour rappel, une directive Européenne est un texte réglementaire rédigé par le conseil de l’Union Européenne (UE) qui définit des objectifs communs pour les états membres. Ces derniers transposent ensuite les exigences de la directive dans le droit national.
Lorsque les DM (Dispositifs Médicaux) répondent aux exigences de la directive, ces derniers se voient apposer le sigle du marquage CE. Il est valable pour les dispositifs fabriqués en Europe comme dans le reste du monde.
De la Directive au Règlement Européen
Après plus de 20 ans d’existence et 5 modifications majeures, la directive 93/42/CEE a été remplacée par un règlement. A la différence des directives européennes qui ont pour objectifs de fixer les objectifs de l’UE tout en laissant aux états membres les moyens de les atteindre (on parle de transposition de la directive dans le droit national), les règlements européens sont obligatoires et directement applicables.
Le nouveau Règlement (UE) 2017/745 du Parlement européen et du Conseil est entré en vigueur le 5 avril 2017. Le règlement intègre désormais 22 règles et 80 critères alors que la directive proposait 18 règles pour 56 critères.
→ Consulter le règlement complet : Règlement (UE) 2017/745
Les défibrillateurs Automatisés Externes (DAE) requalifiés en classe III
Pour rappel, un défibrillateur automatisé externe est un appareil destiné à être utilisé par le grand public en cas d’arrêt cardio-respiratoire. Le fonctionnement du défibrillateur est simple : il analyse le rythme cardiaque du patient puis délivre un choc électrique si le cœur est en fibrillation ou tachycardie ventriculaire. Depuis le décret du 04 mai 2007, toute personne non-médecin est autorisée à utiliser un défibrillateur automatisé externe (DAE).
Anciennement classifiés en IIB, les Défibrillateurs Automatisés Externes (DAE) sont désormais classifiés en classe III (2017). Cela signifie qu’ils font parties des appareils ayant un degré de risque très élevés.
La classification des Dispositifs Médicaux (DM)
Les dispositifs médicaux sont répartis en 4 classes de risque. C’est l’annexe IX de la directive 93/42 définit les règles de classification.
La classification des DM est intrinsèquement liée à leur dangerosité :
- Classe I : faible risque
- Classe IIa : risque moyen
- Classe IIb : risque élevé
- Classe III : degré très élevé de risque
La classification est utilisée en Europe pour définir les exigences réglementaires applicables à un dispositif médical. Autrement dit, plus la classe et élevée, plus les exigences sont élevées (donc plus contraignant pour les fabricants).
La liste des dispositif médicaux est communiqué sur le site de l’ANSM à l’adresse suivante : Liste ANSM des dispositif médicaux
Vous pouvez également utiliser le site eqms.io qui facilite grandement la recherche de DM.
Pourquoi requalifier les défibrillateurs en classe III ?
La question légitime que l’on peut se poser est de savoir pourquoi cette requalification. En effet, les défibrillateurs sont réputés simple d’utilisation et sans risque pour l’utilisateur. D’après nos investigations, la requalification permettrait d’assainir le marché en évitant que des entreprises surfent sur la vague du « défibrillateur » sans avoir les compétences médicales suffisantes. Cela pourrait également venir de la volonté de l’UE d’harmoniser sa réglementation en matière de DM avec celle de la FDA aux Etats-Unis.
La classe III : quels impacts pour les DAE et les fabricants ?
Le marquage CE pour « Conformité Européenne » est le marquage réglementaire qui atteste que le produit est conforme aux exigences réglementaires et donc qu’il peut être commercialisé. Pour obtenir le marquage CE, les entreprises doivent réaliser des contrôles et essais qui prouvent la conformité de leurs produits. Pour cela, elles font appel à un Organisme Notifié (ON) qui est en charge de l’évaluation de conformité (sauf DM de classe I).
Dans le cadre des dispositifs médicaux les exigences réglementaires varient selon la classe. Les procédures possibles à appliquer selon la classe du dispositif sont définies dans les annexes de la directive 93/42CEE :
- Annexe II : Déclaration CE de conformité (système complet d’assurance de la qualité)
- Annexe III : Examen CE de type (Attestation par l’organisme notifié qu’un échantillon représentatif de la production satisfait aux exigences essentielles)
- Annexe IV : Vérification CE
- Annexe V : Déclaration CE de conformité (assurance de la qualité de la production)
- Annexe VI : Déclaration CE de conformité (assurance de la qualité des produits)
- Annexe VII : Déclaration CE de conformité sans recours à un organisme habilité
Ainsi, l’évaluation de la conformité d’un DM de classe I (Annexe VII) repose sur une auto-déclaration du fabricant puisqu’il n’y a pas de recours à un organisme notifié vu le faible degré de vulnérabilité de ces produits. Le fabricant produit un dossier technique et met en place les procédures obligatoires pour entrer en conformité avec la réglementation.
Concernant les dispositifs médicaux de classe IIa (Annexe VII et Annexe IV ou Annexe V ou Annexe VI) et IIb (Annexe III et Annexe IV ou Annexe V ou Annexe VI), le fabricant produit un dossier technique et met en place les processus obligatoire comme vu précédemment. Néanmoins, la mise en place d’un système complet d’assurance qualité contrôlé par un organisme notifié est nécessaire. Il s’agit dès lors d’une procédure beaucoup plus lourde et complexe. Cette procédure peut peut se formaliser par le biais de la certification ISO 13485. Cette certification est l’équivalent de l’ISO9001 adapté au dispositifs médicaux (traçabilité et matériovigilance).
L’ISO 13485 énonce les exigences relatives au système de management de la qualité lorsqu’un organisme doit démontrer son aptitude à fournir régulièrement des dispositifs médicaux et des services associés conformes aux exigences des clients et aux exigences réglementaires applicables
Pour en savoir plus : www.iso.org
La classe IIb de la classe IIa se distingue par le fait que l’organisme certificateur va prélever un échantillon de la production et vérifier sa conformité au dossier technique par le biais de tests techniques.
Le marquage en DM de classe III (Annexe II), est la procédure la plus complexe. La fabricant doit établir le dossier technique, mettre en place un système de management de la qualité total. L’organisme certificateur procédera ensuite à un examen de la conception.
A noter que le règlement 2017/745 précise que, dans le cas des dispositifs implantables et des dispositifs de classe III, des investigations cliniques (IC) doivent être conduites, sauf si les trois critères suivants sont respectés :
- Le dispositif a été conçu en modifiant un dispositif déjà commercialisé par le même fabricant ;
- L’équivalence avec ce dispositif est démontrée et approuvée par l’ON
- L’évaluation clinique du dispositif actuellement commercialisé suffit à démontrer la conformité du dispositif modifié avec les exigences pertinentes en matière de sécurité et de performance
Voici également un tableau publié par l’HAS (Haute Autorité de Santé) qui récapitule la procédure d’évaluation de la conformité en fonction de la classe du dispositif médical :
Classe III et maintenance des défibrillateurs automatisés externes
Le changement de classification ne changera pas pour autant les procédures de maintenance des défibrillateurs automatisés externes. Le changement de classe implique uniquement des contraintes pour l’obtention du marquage CE pour qu’un DM puisse être commercialisé.
L’ANSM le stipule bien dans son guide de 2011 intitulé Mise au point sur la maintenance des dispositifs médicaux : « Les dispositions réglementaires relatives à la maintenance des dispositifs médicaux prévues par le Code de la santé publique concernent la phase d’exploitation, c’est-à-dire la phase après la mise sur le marché. »
La liste des dispositifs médicaux soumis à l’obligation de maintenance est d’ailleurs définie par l’arrêté du 3 mars 2003.
Calendrier du nouveau règlement européen
À compter du 26 mai 2020, toute publication d’une notification relative à un organisme notifié conformément aux directives 90/385/CEE et 93/42/CEE est invalidée.
Les certificats délivrés par des organismes notifiés conformément aux directives 90/385/CEE et 93/42/CEE avant le 25 mai 2017 conservent leur validité jusqu’à la fin de la période indiquée sur ces certificats, sauf pour les certificats délivrés conformément à l’annexe IV de la directive 90/385/CEE ou à l’annexe IV de la directive 93/42/CEE, qui sont invalidés au plus tard le 27 mai 2022.
Les certificats délivrés par des organismes notifiés conformément aux directives 90/385/CEE et 93/42/CEE à partir du 25 mai 2017 restent valables jusqu’à la fin de la période indiquée sur le certificat, laquelle n’excède pas cinq ans après la délivrance dudit certificat. Ils sont toutefois invalidés au plus tard le 27 mai 2024.
Note importante : Les informations fournies dans cet article sont à titre informatif uniquement et ne remplacent en aucun cas l'avis, le diagnostic ou le traitement d'un médecin ou d'un professionnel de santé qualifié. Avant de prendre toute décision concernant votre santé, veuillez consulter un professionnel de santé qualifié.